How to extract a subnetwork that revolves around a specific gene of interest?
Notes:
# To answer this question, without loss of generality we formulate it as an often-encountered problem: from the whole human gene network, how to derive a subnetwork that revolves around a gene 'NANOG' and its direct interacting neighbors.
# 1) load the whole human gene network. The network is extracted from the STRING database (version 9.1). Only those associations with medium confidence (score>=400) are retained
network <- dRDataLoader(RData = "org.Hs.string")
'org.Hs.string' (from https://github.com/hfang-bristol/RDataCentre/blob/master/dnet/1.0.7/org.Hs.string.RData?raw=true) has been loaded into the working environment (at 2017-03-27 20:08:43)
# If required, can only focus on those edges with confidence score >= 900
network <- igraph::subgraph.edges(network, eids=E(network)[combined_score>=900])
network
IGRAPH UN-- 11046 213579 --
+ attr: name (v/c), seqid (v/c), geneid (v/n), symbol (v/c),
| description (v/c), neighborhood_score (e/n), fusion_score (e/n),
| cooccurence_score (e/n), coexpression_score (e/n), experimental_score
| (e/n), database_score (e/n), textmining_score (e/n), combined_score
| (e/n)
+ edges (vertex names):
[1] 3021157--3031351 3028171--3031351 3017550--3029275 3017550--3027007
[5] 3017550--3021301 3017550--3030437 3014534--3017550 3017550--3028725
[9] 3016736--3017550 3017550--3029563 3017550--3032559 3017550--3021463
[13] 3014981--3025139 3014981--3018212 3014981--3028439 3014981--3033369
+ ... omitted several edges
# 2) find the gene 'NANOG' and its direct neighbors
ind <- match(V(network)$symbol, 'NANOG')
nodes_query <- V(network)$name[!is.na(ind)]
g <- dNetInduce(g=network, nodes_query=nodes_query, knn=1, remove.loops=F, largest.comp=T)
g
IGRAPH UN-- 41 148 --
+ attr: name (v/c), seqid (v/c), geneid (v/n), symbol (v/c),
| description (v/c), neighborhood_score (e/n), fusion_score (e/n),
| cooccurence_score (e/n), coexpression_score (e/n), experimental_score
| (e/n), database_score (e/n), textmining_score (e/n), combined_score
| (e/n)
+ edges (vertex names):
[1] 3014314--3016028 3014314--3017025 3014314--3020782 3014351--3014699
[5] 3014351--3017025 3014351--3020782 3014351--3023492 3014652--3014699
[9] 3014652--3018951 3014652--3020782 3014652--3023492 3014652--3025830
[13] 3014652--3031207 3014699--3018228 3014699--3018951 3014699--3020271
+ ... omitted several edges
# 3) visualise the extracted subnetwork
visNet(g, vertex.label=V(g)$symbol, vertex.shape="sphere")
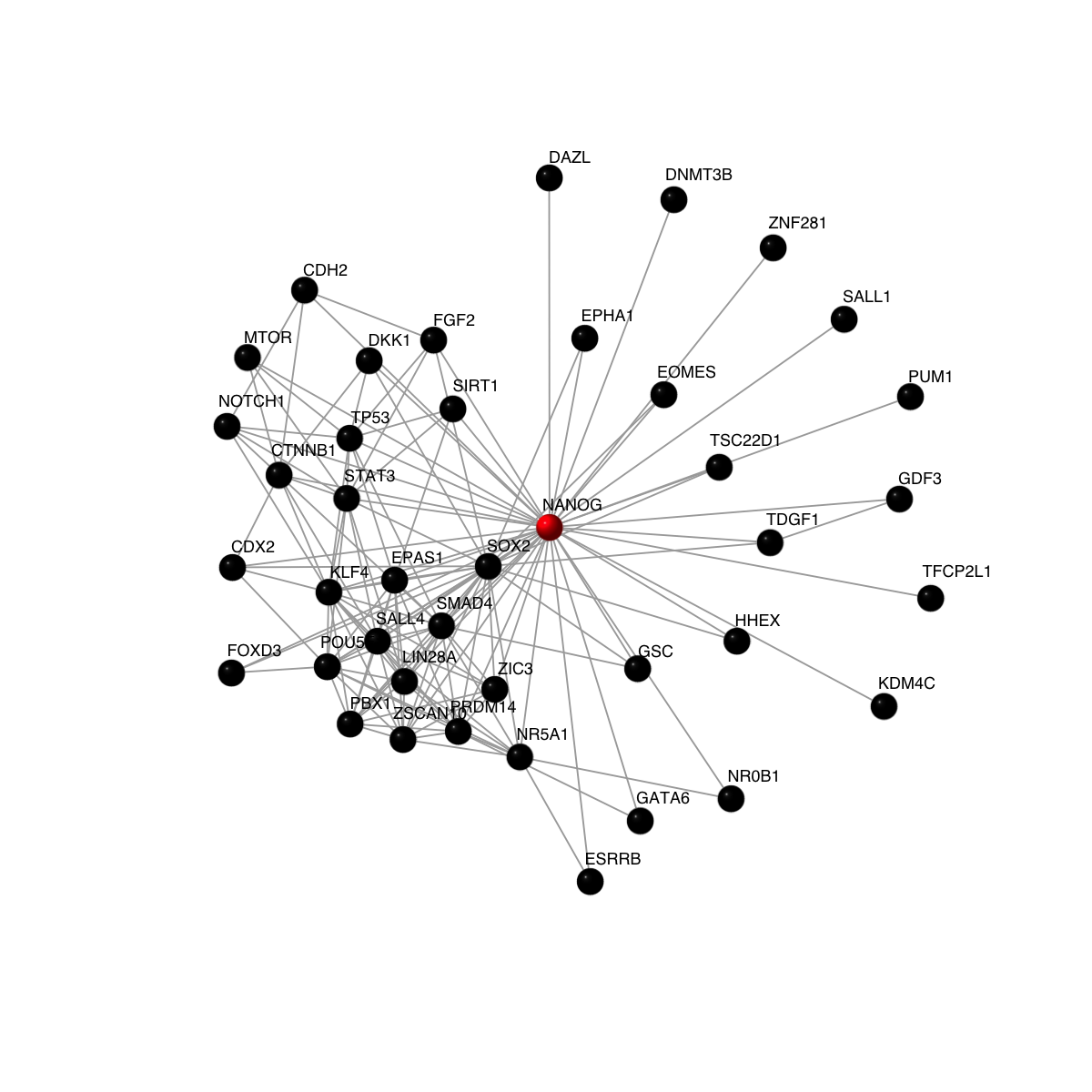
# 4) highlight the query gene in red, and its neighours in black
vcolors <- rep('black', vcount(g))
 vcolors[match('NANOG', V(g)$symbol)] <- 'red'
visNet(g, vertex.label=V(g)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# 5) output the detailed information for genes in the subnetwork onto the file called 'NANOG.subnetwork.txt'
out <- data.frame(GeneID=V(g)$geneid, Symbol=V(g)$symbol, Description=V(g)$description)
vcolors[match('NANOG', V(g)$symbol)] <- 'red'
visNet(g, vertex.label=V(g)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# 5) output the detailed information for genes in the subnetwork onto the file called 'NANOG.subnetwork.txt'
out <- data.frame(GeneID=V(g)$geneid, Symbol=V(g)$symbol, Description=V(g)$description)
 write.table(out, file="NANOG.subnetwork.txt", quote=F, row.names=F,col.names=T,sep="\t")
out
GeneID Symbol
1 27022 FOXD3
2 22943 DKK1
3 4851 NOTCH1
4 1499 CTNNB1
5 79727 LIN28A
6 6997 TDGF1
7 5460 POU5F1
8 63978 PRDM14
9 6657 SOX2
10 5087 PBX1
11 2103 ESRRB
12 4089 SMAD4
13 1000 CDH2
14 2034 EPAS1
15 2041 EPHA1
16 2516 NR5A1
17 23411 SIRT1
18 57167 SALL4
19 29842 TFCP2L1
20 79923 NANOG
21 23081 KDM4C
22 1789 DNMT3B
23 8848 TSC22D1
24 6299 SALL1
25 2627 GATA6
26 1045 CDX2
27 7157 TP53
28 2247 FGF2
29 6774 STAT3
30 3087 HHEX
31 9573 GDF3
32 190 NR0B1
33 8320 EOMES
34 9698 PUM1
35 9314 KLF4
36 23528 ZNF281
37 84891 ZSCAN10
38 7547 ZIC3
39 145258 GSC
40 2475 MTOR
41 1618 DAZL
Description
1 forkhead box D3
2 dickkopf WNT signaling pathway inhibitor 1
3 notch 1
4 catenin (cadherin-associated protein), beta 1, 88kDa
5 lin-28 homolog A (C. elegans)
6 teratocarcinoma-derived growth factor 1
7 POU class 5 homeobox 1
8 PR domain containing 14
9 SRY (sex determining region Y)-box 2
10 pre-B-cell leukemia homeobox 1
11 estrogen-related receptor beta
12 SMAD family member 4
13 cadherin 2, type 1, N-cadherin (neuronal)
14 endothelial PAS domain protein 1
15 EPH receptor A1
16 nuclear receptor subfamily 5, group A, member 1
17 sirtuin 1
18 spalt-like transcription factor 4
19 transcription factor CP2-like 1
20 Nanog homeobox
21 lysine (K)-specific demethylase 4C
22 DNA (cytosine-5-)-methyltransferase 3 beta
23 TSC22 domain family, member 1
24 spalt-like transcription factor 1
25 GATA binding protein 6
26 caudal type homeobox 2
27 tumor protein p53
28 fibroblast growth factor 2 (basic)
29 signal transducer and activator of transcription 3 (acute-phase response factor)
30 hematopoietically expressed homeobox
31 growth differentiation factor 3
32 nuclear receptor subfamily 0, group B, member 1
33 eomesodermin
34 pumilio RNA-binding family member 1
35 Kruppel-like factor 4 (gut)
36 zinc finger protein 281
37 zinc finger and SCAN domain containing 10
38 Zic family member 3
39 goosecoid homeobox
40 mechanistic target of rapamycin (serine/threonine kinase)
41 deleted in azoospermia-like
# A more complicated question could be: the interaction neighbors are also required to be functionally related to 'stem'. For this, we can further use terms from Gene Ontology (and its subontology: Biological Process).
keyword <- 'stem'
org.Hs.egGOBP <- dRDataLoader(RData = "org.Hs.egGOBP")
'org.Hs.egGOBP' (from https://github.com/hfang-bristol/RDataCentre/blob/master/dnet/1.0.7/org.Hs.egGOBP.RData?raw=true) has been loaded into the working environment (at 2017-03-27 20:08:49)
ind <- grep(keyword, org.Hs.egGOBP$set_info$name, ignore.case=T, perl=T)
genes <- unique(unlist(org.Hs.egGOBP$gs[ind]))
# Next, identify genes in the subnetwork that are functionally related to 'stem'
ind <- match(V(g)$geneid, genes)
intersect_genes <- V(g)$symbol[!is.na(ind)]
intersect_genes
[1] "FOXD3" "NOTCH1" "CTNNB1" "LIN28A" "TDGF1" "POU5F1" "PRDM14"
[8] "SOX2" "PBX1" "ESRRB" "SMAD4" "CDH2" "EPAS1" "EPHA1"
[15] "SALL4" "NANOG" "CDX2" "FGF2" "STAT3" "GDF3" "EOMES"
[22] "PUM1" "KLF4" "ZNF281" "ZSCAN10" "ZIC3" "GSC"
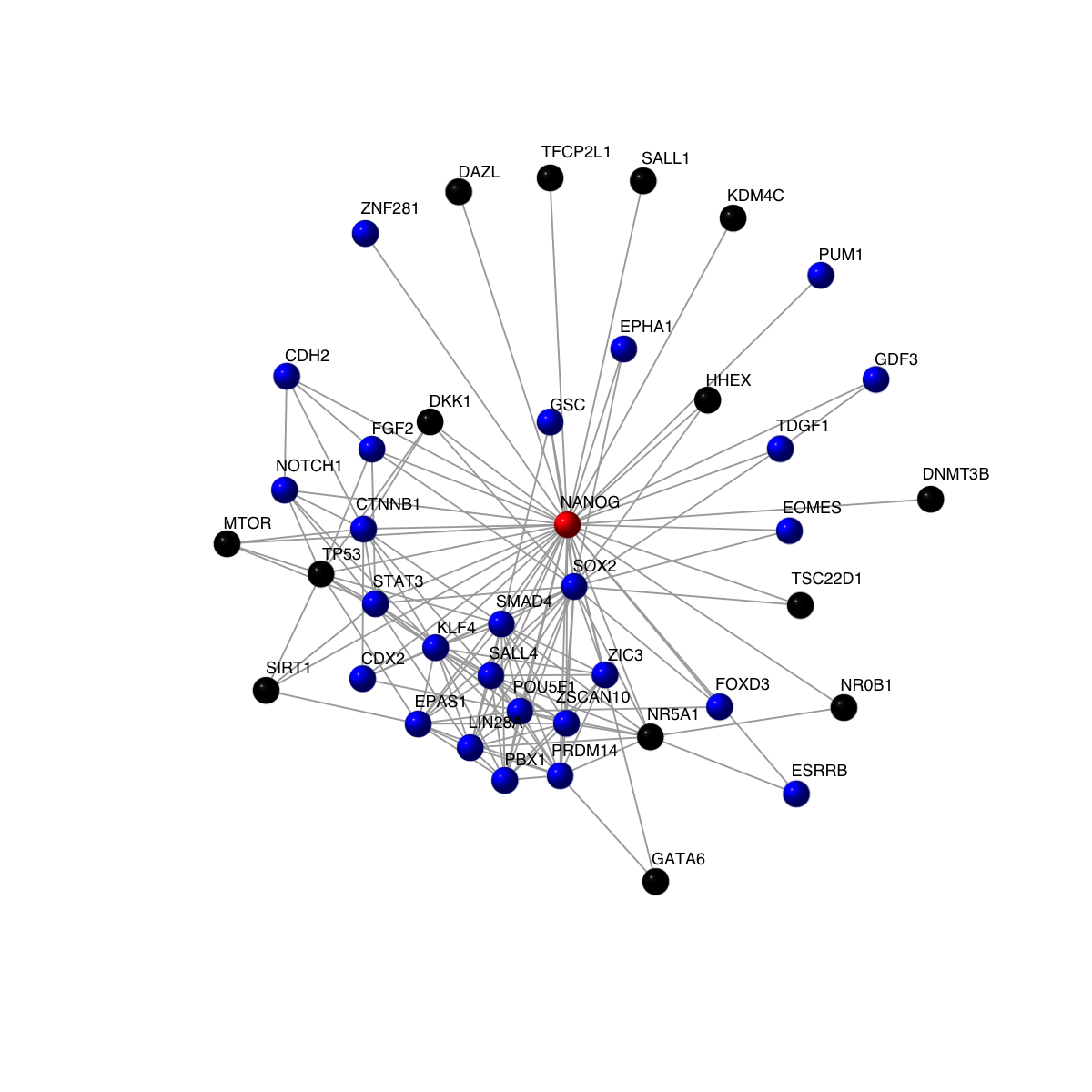
# Now, highlight the query gene in red, its 'stem'-related neighours in blue, and the rest in black
vcolors <- rep('black', vcount(g))
vcolors[match(intersect_genes, V(g)$symbol)] <- 'blue'
vcolors[match('NANOG', V(g)$symbol)] <- 'red'
visNet(g, vertex.label=V(g)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# Alternatively, remain only genes related to 'stem' and the query gene 'NANOG'.
both_genes <- c(intersect_genes, 'NANOG')
write.table(out, file="NANOG.subnetwork.txt", quote=F, row.names=F,col.names=T,sep="\t")
out
GeneID Symbol
1 27022 FOXD3
2 22943 DKK1
3 4851 NOTCH1
4 1499 CTNNB1
5 79727 LIN28A
6 6997 TDGF1
7 5460 POU5F1
8 63978 PRDM14
9 6657 SOX2
10 5087 PBX1
11 2103 ESRRB
12 4089 SMAD4
13 1000 CDH2
14 2034 EPAS1
15 2041 EPHA1
16 2516 NR5A1
17 23411 SIRT1
18 57167 SALL4
19 29842 TFCP2L1
20 79923 NANOG
21 23081 KDM4C
22 1789 DNMT3B
23 8848 TSC22D1
24 6299 SALL1
25 2627 GATA6
26 1045 CDX2
27 7157 TP53
28 2247 FGF2
29 6774 STAT3
30 3087 HHEX
31 9573 GDF3
32 190 NR0B1
33 8320 EOMES
34 9698 PUM1
35 9314 KLF4
36 23528 ZNF281
37 84891 ZSCAN10
38 7547 ZIC3
39 145258 GSC
40 2475 MTOR
41 1618 DAZL
Description
1 forkhead box D3
2 dickkopf WNT signaling pathway inhibitor 1
3 notch 1
4 catenin (cadherin-associated protein), beta 1, 88kDa
5 lin-28 homolog A (C. elegans)
6 teratocarcinoma-derived growth factor 1
7 POU class 5 homeobox 1
8 PR domain containing 14
9 SRY (sex determining region Y)-box 2
10 pre-B-cell leukemia homeobox 1
11 estrogen-related receptor beta
12 SMAD family member 4
13 cadherin 2, type 1, N-cadherin (neuronal)
14 endothelial PAS domain protein 1
15 EPH receptor A1
16 nuclear receptor subfamily 5, group A, member 1
17 sirtuin 1
18 spalt-like transcription factor 4
19 transcription factor CP2-like 1
20 Nanog homeobox
21 lysine (K)-specific demethylase 4C
22 DNA (cytosine-5-)-methyltransferase 3 beta
23 TSC22 domain family, member 1
24 spalt-like transcription factor 1
25 GATA binding protein 6
26 caudal type homeobox 2
27 tumor protein p53
28 fibroblast growth factor 2 (basic)
29 signal transducer and activator of transcription 3 (acute-phase response factor)
30 hematopoietically expressed homeobox
31 growth differentiation factor 3
32 nuclear receptor subfamily 0, group B, member 1
33 eomesodermin
34 pumilio RNA-binding family member 1
35 Kruppel-like factor 4 (gut)
36 zinc finger protein 281
37 zinc finger and SCAN domain containing 10
38 Zic family member 3
39 goosecoid homeobox
40 mechanistic target of rapamycin (serine/threonine kinase)
41 deleted in azoospermia-like
# A more complicated question could be: the interaction neighbors are also required to be functionally related to 'stem'. For this, we can further use terms from Gene Ontology (and its subontology: Biological Process).
keyword <- 'stem'
org.Hs.egGOBP <- dRDataLoader(RData = "org.Hs.egGOBP")
'org.Hs.egGOBP' (from https://github.com/hfang-bristol/RDataCentre/blob/master/dnet/1.0.7/org.Hs.egGOBP.RData?raw=true) has been loaded into the working environment (at 2017-03-27 20:08:49)
ind <- grep(keyword, org.Hs.egGOBP$set_info$name, ignore.case=T, perl=T)
genes <- unique(unlist(org.Hs.egGOBP$gs[ind]))
# Next, identify genes in the subnetwork that are functionally related to 'stem'
ind <- match(V(g)$geneid, genes)
intersect_genes <- V(g)$symbol[!is.na(ind)]
intersect_genes
[1] "FOXD3" "NOTCH1" "CTNNB1" "LIN28A" "TDGF1" "POU5F1" "PRDM14"
[8] "SOX2" "PBX1" "ESRRB" "SMAD4" "CDH2" "EPAS1" "EPHA1"
[15] "SALL4" "NANOG" "CDX2" "FGF2" "STAT3" "GDF3" "EOMES"
[22] "PUM1" "KLF4" "ZNF281" "ZSCAN10" "ZIC3" "GSC"
# Now, highlight the query gene in red, its 'stem'-related neighours in blue, and the rest in black
vcolors <- rep('black', vcount(g))
vcolors[match(intersect_genes, V(g)$symbol)] <- 'blue'
vcolors[match('NANOG', V(g)$symbol)] <- 'red'
visNet(g, vertex.label=V(g)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# Alternatively, remain only genes related to 'stem' and the query gene 'NANOG'.
both_genes <- c(intersect_genes, 'NANOG')
 both_genes
[1] "FOXD3" "NOTCH1" "CTNNB1" "LIN28A" "TDGF1" "POU5F1" "PRDM14"
[8] "SOX2" "PBX1" "ESRRB" "SMAD4" "CDH2" "EPAS1" "EPHA1"
[15] "SALL4" "NANOG" "CDX2" "FGF2" "STAT3" "GDF3" "EOMES"
[22] "PUM1" "KLF4" "ZNF281" "ZSCAN10" "ZIC3" "GSC" "NANOG"
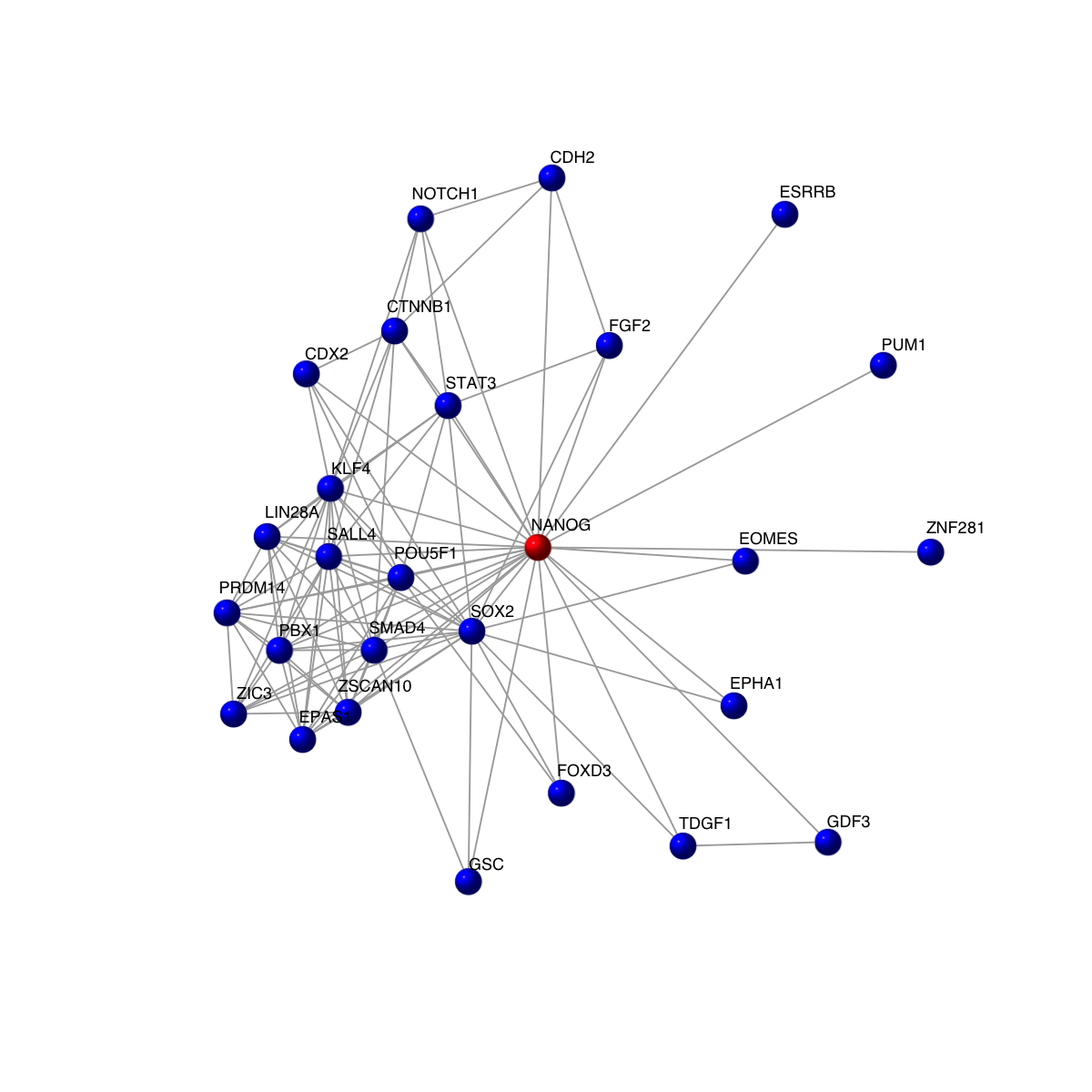
# extract the subnetwork containing the gene 'CDKN2D' and its differentiation-related neighbors
ind <- match(V(g)$symbol, both_genes)
nodes_query <- V(g)$name[!is.na(ind)]
g1 <- dNetInduce(g, nodes_query=nodes_query, knn=0, remove.loops=F, largest.comp=T)
vcolors <- rep('blue', vcount(g1))
vcolors[match('NANOG', V(g1)$symbol)] <- 'red'
visNet(g1, vertex.label=V(g1)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# Also, only edges linked to the query gene 'NANOG' are remained.
ind <- match('NANOG', V(g1)$symbol)
both_genes
[1] "FOXD3" "NOTCH1" "CTNNB1" "LIN28A" "TDGF1" "POU5F1" "PRDM14"
[8] "SOX2" "PBX1" "ESRRB" "SMAD4" "CDH2" "EPAS1" "EPHA1"
[15] "SALL4" "NANOG" "CDX2" "FGF2" "STAT3" "GDF3" "EOMES"
[22] "PUM1" "KLF4" "ZNF281" "ZSCAN10" "ZIC3" "GSC" "NANOG"
# extract the subnetwork containing the gene 'CDKN2D' and its differentiation-related neighbors
ind <- match(V(g)$symbol, both_genes)
nodes_query <- V(g)$name[!is.na(ind)]
g1 <- dNetInduce(g, nodes_query=nodes_query, knn=0, remove.loops=F, largest.comp=T)
vcolors <- rep('blue', vcount(g1))
vcolors[match('NANOG', V(g1)$symbol)] <- 'red'
visNet(g1, vertex.label=V(g1)$symbol, vertex.shape="sphere", vertex.color=vcolors)
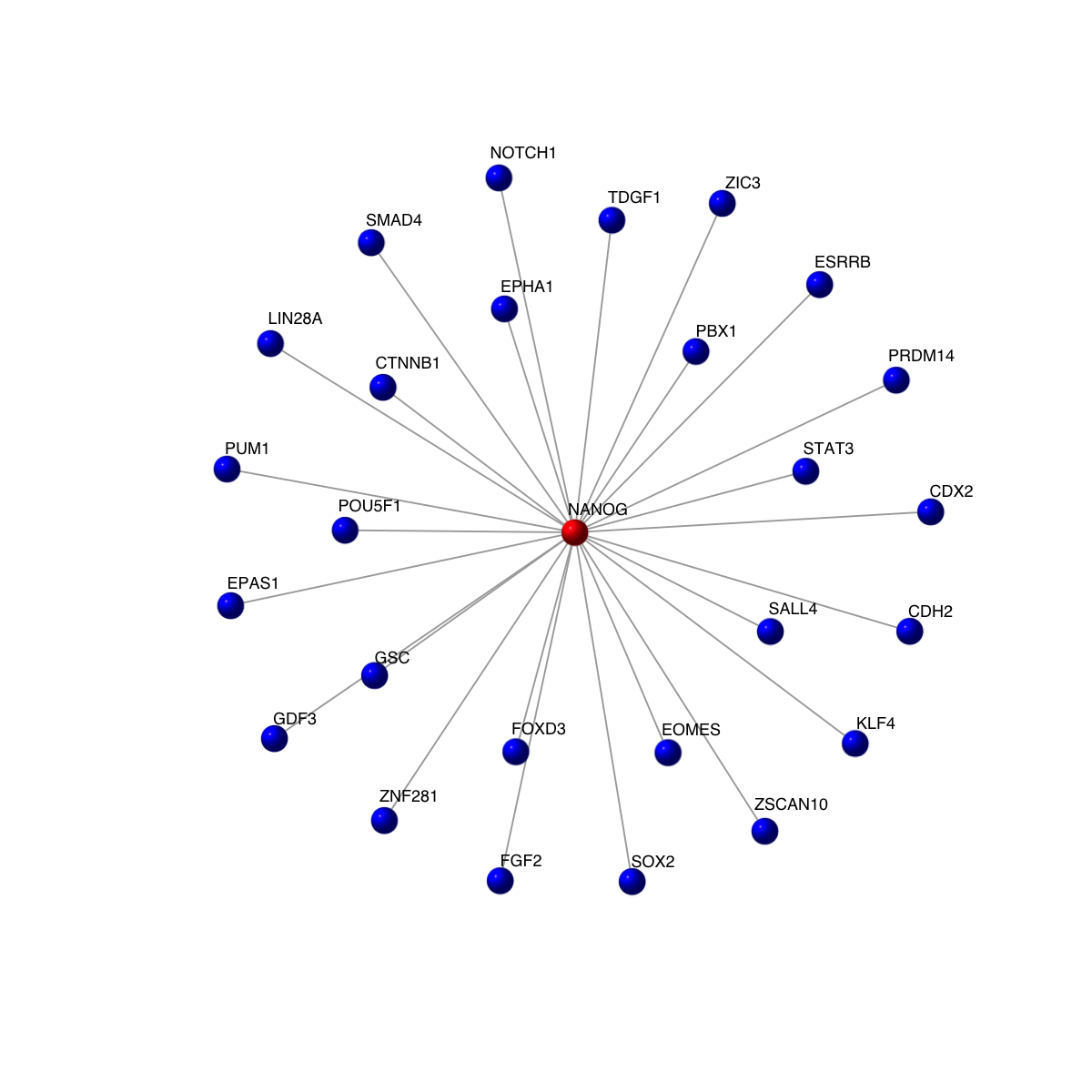
# Also, only edges linked to the query gene 'NANOG' are remained.
ind <- match('NANOG', V(g1)$symbol)
 eids <- E(g1)[from(ind)]
g2 <- subgraph.edges(g1, eids=eids)
vcolors <- rep('blue', vcount(g2))
vcolors[match('NANOG', V(g2)$symbol)] <- 'red'
visNet(g2, vertex.label=V(g2)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# In addition to the direct interacting neighbors, the indirect neighbors can be specifed via the parameter 'knn' in the function dNetInduce.
eids <- E(g1)[from(ind)]
g2 <- subgraph.edges(g1, eids=eids)
vcolors <- rep('blue', vcount(g2))
vcolors[match('NANOG', V(g2)$symbol)] <- 'red'
visNet(g2, vertex.label=V(g2)$symbol, vertex.shape="sphere", vertex.color=vcolors)
# In addition to the direct interacting neighbors, the indirect neighbors can be specifed via the parameter 'knn' in the function dNetInduce.
)
)
)
)
)