# Prior to this question, please allow us to talk about the procedure of how to identify gene-active networks/subgraphs.
The dnet package takes as input a list of genes with the significance information (p-values or FDR), and superposes these genes onto a gene interaction network. From this network with imposed node information, dnet sets up the pipeline to search for a maximum-scoring subgraph. Given the threshold of tolerable p-value, it gives positive scores for nodes with p-values below the threshold (nodes of interest), and negative scores for nodes with threshold-above p-values (intolerable). After score transformation, the search for a maximum scoring subgraph is deduced to find the connected subgraph that is enriched with positive-score nodes, allowing for a few negative-score nodes as linkers. This objective is met through minimum spanning tree finding and post-processing. The solver is deterministic, only determined by the given tolerable p-value threshold. For identification of the subgraph with a desired number of nodes, an iterative procedure is also developed to fine-tune tolerable thresholds. This pipeline procedure is implemented in the function
dNetPipeline.
#
The calculation of gene significance depends on the nature of input data (especially samples under consideration). It can be: 1) samples that are grouped, 2) samples collected as a time series, and 3) samples in a clinical setting (with survival data).
# For
grouped samples, the the p-value significance for genes can be calculated using the package 'limma' which is widely used for idnetification of differentially expressed genes. The example for this can be found in the
Demo for multilayer replication timing dataset page.
# For
time-course samples, the dnet package has developed a singular value decomposition-based method to obtain gene significance (see
dSVDsignif). The example for this can be found in the
Demo for time-course expression dataset page.
# For
survival samples, survival analysis should be applied using Cox proportional hazards model, from which Cox hazard ratio and associated p-value for each gene can be calculated. This can be done via the 'survival' package, and is demonstrated in the
Demo for TCGA mutation and survival dataset page.
###########################################################################
# Below we will use human embryo dataset from Fang et al to show how to calculate the gene signficance. This involves six successive developmental stages (S9-S14) with three replicates (R1-R3) for each stage, including:
## Fang: an expression matrix of 5,441 genes/probesets X 18 samples;
## Fang.geneinfo: a matrix of 5,441 X 3 containing gene/probeset information;
## Fang.sampleinfo: a matrix of 18 X 3 containing sample information.
###########################################################################
## import data containing three variables ('Fang', 'Fang.geneinfo' and 'Fang.sampleinfo')
data(Fang)
data <- Fang
fdata <- as.data.frame(Fang.geneinfo[,2:3])
rownames(fdata) <- Fang.geneinfo[,1]
pdata <- as.data.frame(Fang.sampleinfo[,2:3])
rownames(pdata) <- Fang.sampleinfo[,1]
## for the ease of managing this dataset, we create the 'eset' object:
colmatch <- match(rownames(pdata),colnames(data))
rowmatch <- match(rownames(fdata),rownames(data))
data <- data[rowmatch,colmatch]
eset <- new("ExpressionSet", exprs=as.matrix(data), phenoData=as(pdata,"AnnotatedDataFrame"), featureData=as(fdata,"AnnotatedDataFrame"))
## Since this dataset is generated by Affymetrix, so data at the probeset level should be converted to data at the gene level. For this, we define a function 'prob2gene' to convert probeset-centric to entrezgene-centric expression levels:
prob2gene <- function(eset){
fdat <- fData(eset)
tmp <- as.matrix(unique(fdat[c("EntrezGene", "Symbol")]))
forder <- tmp[order(as.numeric(tmp[,1])),]
forder <- forder[!is.na(forder[,1]),]
rownames(forder) <- forder[,2]
system.time({
dat <- exprs(eset)
edat <- matrix(data=NA, nrow=nrow(forder), ncol=ncol(dat))
for (i in 1:nrow(forder)){
ind <- which(fdat$EntrezGene==as.numeric(forder[i,1]))
if (length(ind) == 1){
edat[i,] <- dat[ind,]
}else{
edat[i,] <- apply(dat[ind,],2,mean)
}
}
})
rownames(edat) <- rownames(forder) # as gene symbols
colnames(edat) <- rownames(pData(eset))
esetGene <- new('ExpressionSet',exprs=data.frame(edat),phenoData=as(pData(eset),"AnnotatedDataFrame"),featureData=as(data.frame(forder),"AnnotatedDataFrame"))
return(esetGene)
}
## 'esetGene' stores the gene-level expression data
esetGene <- prob2gene(eset)
esetGene
ExpressionSet (storageMode: lockedEnvironment)
assayData: 4139 features, 18 samples
element names: exprs
protocolData: none
phenoData
sampleNames: S9_R1 S9_R2 ... S14_R3 (18 total)
varLabels: Stage Replicate
varMetadata: labelDescription
featureData
featureNames: A2M AADAC ... LOC100131613 (4139 total)
fvarLabels: EntrezGene Symbol
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation:
S10_S9 S11_S10 S12_S11 S13_S12 S14_S13
994 215 165 204 330
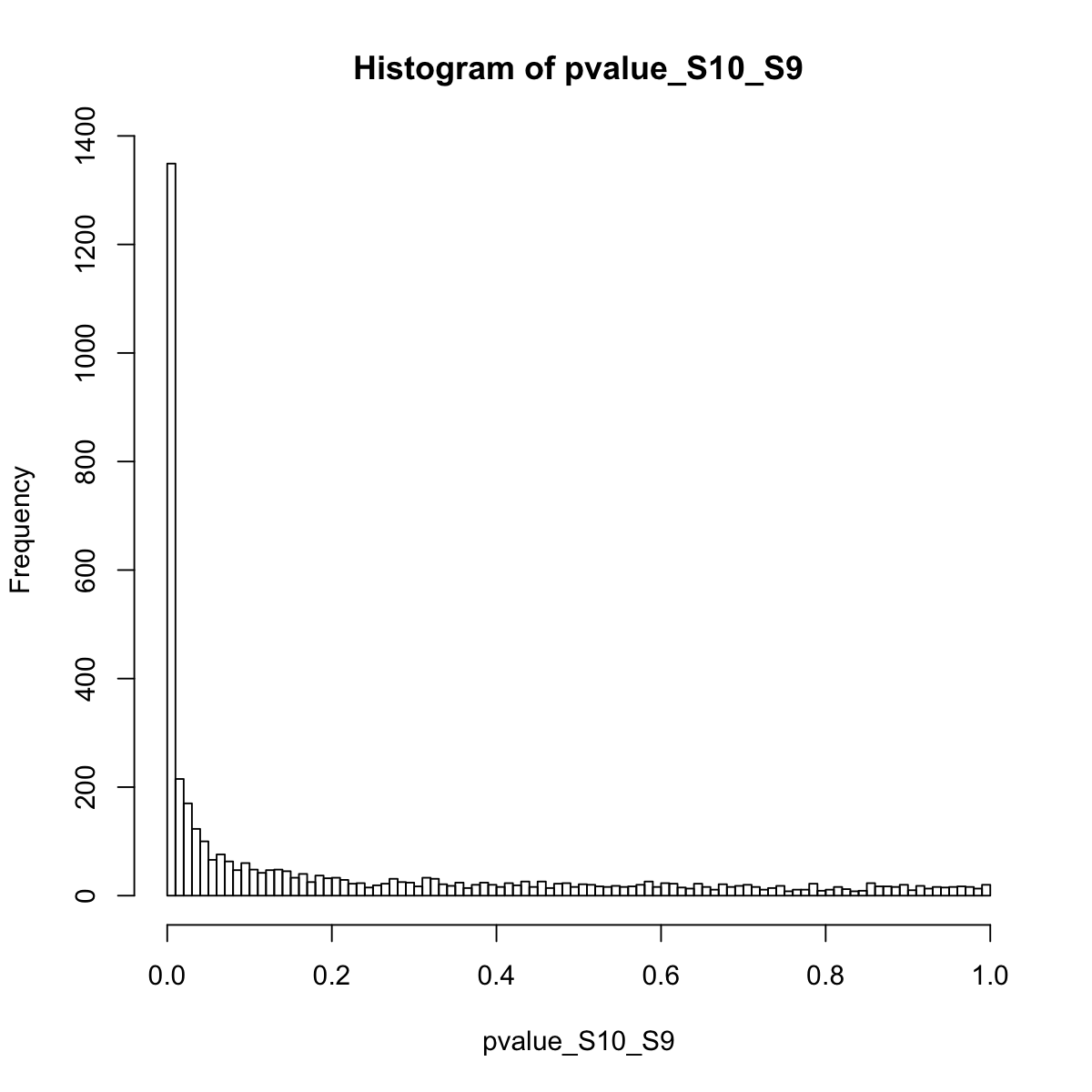
### extract gene signficance according to two sample groups (S10 versus S9):
pvalue_S10_S9 <- pvals[,1]
hist(pvalue_S10_S9, 100)
 Start at 2017-03-27 20:06:51
First, singular value decomposition of the input matrix (with 4139 rows and 18 columns)...
Second, determinate the eigens...
via automatically deciding on the number of dominant eigens under the cutoff of 1.00e-02 pvalue
number of the eigens in consideration: 2
Third, construct the gene-specific projection vector,and calculate distance statistics...
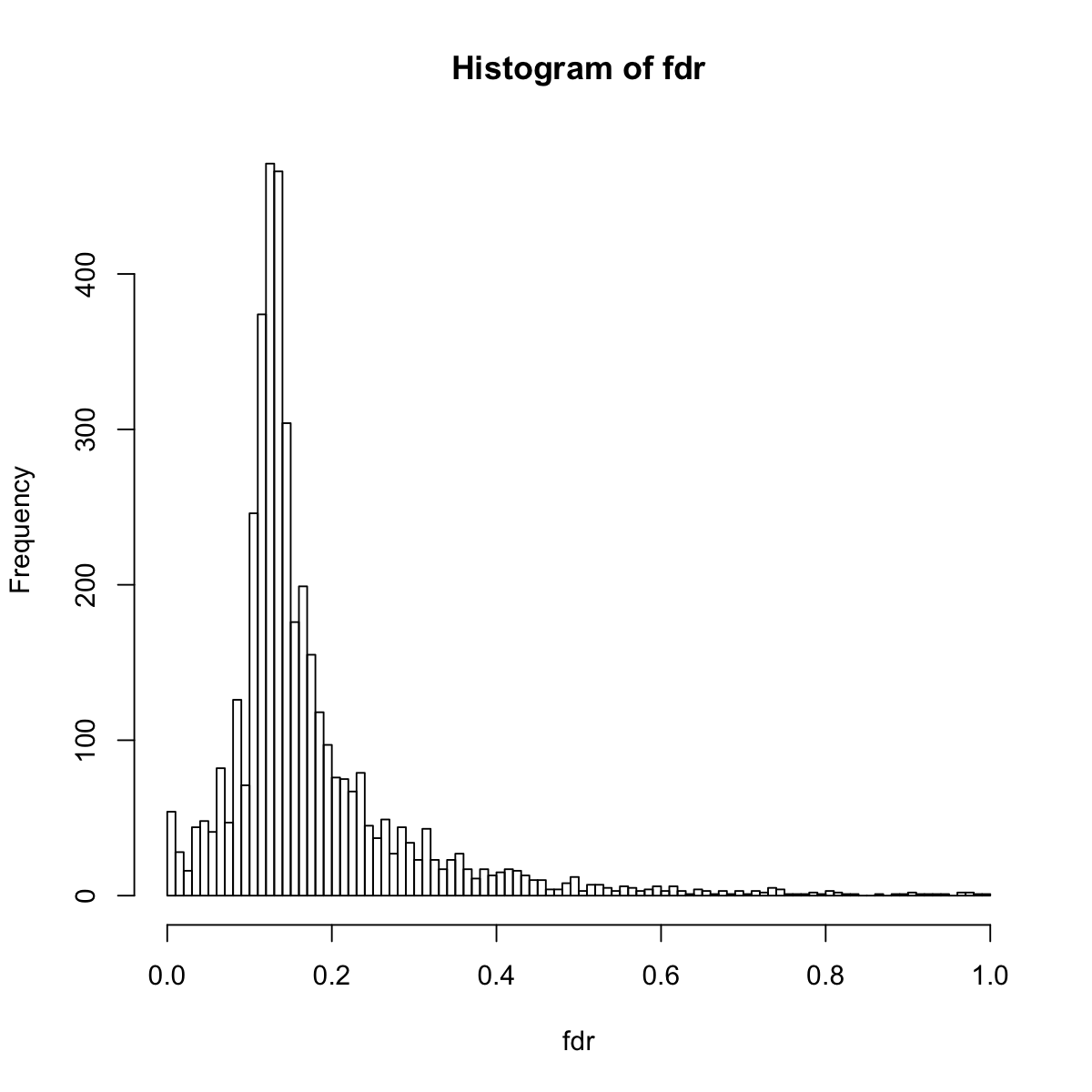
Finally, obtain gene significance (fdr) based on 100 permutations...
doing row-wise permutations...
estimating fdr...
1 (out of 4139) at 2017-03-27 20:07:02
414 (out of 4139) at 2017-03-27 20:07:09
828 (out of 4139) at 2017-03-27 20:07:17
1242 (out of 4139) at 2017-03-27 20:07:25
1656 (out of 4139) at 2017-03-27 20:07:33
2070 (out of 4139) at 2017-03-27 20:07:41
2484 (out of 4139) at 2017-03-27 20:07:48
2898 (out of 4139) at 2017-03-27 20:07:55
3312 (out of 4139) at 2017-03-27 20:08:02
3726 (out of 4139) at 2017-03-27 20:08:08
4139 (out of 4139) at 2017-03-27 20:08:14
using stepup procedure...
Finish at 2017-03-27 20:08:14
Runtime in total is: 83 secs
Start at 2017-03-27 20:06:51
First, singular value decomposition of the input matrix (with 4139 rows and 18 columns)...
Second, determinate the eigens...
via automatically deciding on the number of dominant eigens under the cutoff of 1.00e-02 pvalue
number of the eigens in consideration: 2
Third, construct the gene-specific projection vector,and calculate distance statistics...
Finally, obtain gene significance (fdr) based on 100 permutations...
doing row-wise permutations...
estimating fdr...
1 (out of 4139) at 2017-03-27 20:07:02
414 (out of 4139) at 2017-03-27 20:07:09
828 (out of 4139) at 2017-03-27 20:07:17
1242 (out of 4139) at 2017-03-27 20:07:25
1656 (out of 4139) at 2017-03-27 20:07:33
2070 (out of 4139) at 2017-03-27 20:07:41
2484 (out of 4139) at 2017-03-27 20:07:48
2898 (out of 4139) at 2017-03-27 20:07:55
3312 (out of 4139) at 2017-03-27 20:08:02
3726 (out of 4139) at 2017-03-27 20:08:08
4139 (out of 4139) at 2017-03-27 20:08:14
using stepup procedure...
Finish at 2017-03-27 20:08:14
Runtime in total is: 83 secs
hist(fdr, 100)
)
)